ALTA ESTATURA
DEFINIÇÃO
ALTA ESTATURA: estatura superior a 2 DP em relação à média para o sexo, idade cronológica e grupo populacional1.
ETIOLOGIA
| ETIOLOGIA | EXAMES | COMENTÁRIOS |
| VARIAÇÃO NORMAL DO CRESCIMENTO | ||
| Alta estatura constitucional, genética ou familiar |
|
|
| DOENÇAS ENDÓCRINO-METABÓLICAS | ||
| Recém-nascido filho de mãe diabética |
|
|
| Hiperinsulinismo neonatal |
|
|
| Gigantismo hipofisário |
|
|
| Acromegaloidismo |
|
|
| Puberdade precoce |
|
|
| Hiperplasia adrenal congênita |
|
|
| Hipogonadismo |
|
|
| Deficiência de aromatase |
|
|
| Resistência estrogênica |
|
|
| Hipertireoidismo |
|
|
| Deficiência familiar de glicocorticóide |
|
|
| Resistência familiar aos glicocorticóides |
|
|
| Obesidade exógena |
|
|
| Homocistinúria |
|
|
| Lipodistrofia congênita total |
|
|
| Trissomia do receptor do IGF-1 (IGF-1R) |
|
|
| SÍNDROMES GENÉTICAS | ||
| Síndrome de Marfan |
|
|
| Síndrome de Loeys-Dietz (SLD) ou síndrome de Marfan tipo II |
|
|
| Síndrome de Beals ou aracnodactilia contratural congênita |
|
|
| Síndrome de Klinefelter |
|
|
| Síndrome XYY, síndrome do duplo Y ou homem 47,XYY |
|
|
| Trissomia X, síndrome do triplo X ou mulher 47,XXX |
|
|
| Síndrome do X frágil |
|
|
| Síndrome de Beckwith-Wiedemann (SBW) |
|
|
| Síndrome de Sotos ou gigantismo cerebral |
|
|
| Síndrome de Weaver |
|
|
| Síndrome de Marshall-Smith |
|
|
| Síndrome de Perlman |
|
|
| Síndrome de Simpson-Golabi-Behmel |
|
|
| Síndrome de Bannayan-Riley-Ruvalcaba |
|
|
| Síndrome de Pallister-Killian |
|
|
| Síndrome de Costello |
|
|
| Síndrome de Partington |
|
|
| Síndrome M-CMTC (macrocefalia, cutis marmorata, telangiectasia congênita) |
|
|
| Neurofibromatose tipo 1 (doença de Von Recklinghausen) |
|
|
| Síndrome de Nevo |
|
|
| Síndrome de Elejalde (displasia acrocefalopolidáctila) |
|
|
| Síndrome CATSHI (camptodactilia, tall stature, scoliosis, hearing loss) |
|
|
DIAGNÓSTICO ETIOLÓGICO DIFERENCIAL DA ALTA ESTATURA
DIAGNÓSTICO ETIOLÓGICO DA ALTA ESTATURA DE INÍCIO PRÉ-NATAL
| HIPOGLICEMIA NEONATAL | PERÍMETRO CEFÁLICO | PRINCIPAIS CARACTERÍSTICAS | DISTÚRBIO |
|---|---|---|---|
| presente (transitória) | normal | Hiperbilirrubinemia, trombose da veia renal, policitemia, cardiomegalia, estenose subaórtica, agenesia lombosacral | RN de mãe diabética |
| presente | normal | Micrognatia, distensão abdominal, visceromegalia | Síndrome de Perlman |
| presente (persistente) | diminuído | Macrossomia | Hiperinsulinismo neonatal |
| presente | aumentado | Macroglossia, visceromegalia, onfalocele, alteração das orelhas, hemi-hipertrofia, displasia renal | Síndrome de Beckwith-Wiedemann |
| presente | aumentado | Macroglossia, visceromegalia, hipotonia, cardiopatia congênita | Síndrome de Simpson-Golabi-Behmel |
| ausente | normal | Cifose, hipotonia muscular, hiperextensibilidade articular | Síndrome de Nevo |
| ausente | normal | Craniossinostose, polidactilia, onfalocele, hiperteilorismo, doença policística renal bilateral, hipoplasia pulmonar e insuficiência pancreática | Síndrome de Elejalde |
| ausente | normal | Polidactilia, fronte proeminente, hiperteilorismo, pescoço curto, cardiopatia congênita, hérnia diafragmática | Síndrome de Pallister-Killian |
| ausente | aumentado | Hiperteilorismo, hipotonia, dificuldade respiratória | Síndrome de Sotos |
| ausente | aumentado | Fronte proeminente, orelhas grandes, micrognatia, hipertricose, cabelos finos, retardo mental, hipoplasia ungueal, camptodactilia | Síndrome de Weaver |
| ausente | aumentado | Fronte proeminente, pseudo-exoftalmia, micrognatia, retardo mental, escleras azuladas | Síndrome de Marshall-Smith |
| Fonte: modificado de ALVES, 2019. | |||
| PADRÃO DA ALTA ESTATURA | PARÃMETRO ADICIONAL | DIAGNÓSTICOS |
|---|---|---|
| proporcional | sobrepeso | Obesidade exógena |
| proporcional | puberdade precoce | Puberdade precoce |
| atraso puberal | Hipogonadismo Mulheres 47, XXX Síndrome de Klinefelter |
|
| proporcional | inteligência normal | Alta estatura constitucional |
| proporcional | déficit intelectual | Síndrome de Sotos Síndrome de Weaver Síndrome de marshall-Smith Síndrome de Beckwith-Wiedemann |
| alta estatura de início ao nascimento | RN filho de mãe diabética Síndromes genéticas |
|
| hipoglicemia neonatal | RN filho de mãe diabética lactentes gigantes Síndrome de Beckwith-Wiedemann Síndrome de Simpson-Golabi-Behmel Síndrome de Perlman |
|
| macrocefalia | Síndrome de Simpson-Golabi-Behmel Síndrome de Sotos Síndrome de Weaver Síndrome de Nevo Síndrome de Marshall-Smith |
|
| alterações renais | Síndrome de Beckwith-Wiedemann Síndrome de Simpson-Golabi-Behmel Síndrome de Sotos Síndrome de Elejalde |
|
| cardiopatias | Síndrome de Marfan Síndrome de Beals Síndrome de Sotos Síndrome de Simpson-Golabi-Behmel Síndrome de Pallister-Killian |
|
| neoplasias | Síndrome de Sotos Síndrome de Weaver Síndrome de Perlman Síndrome de Simpson-Golabi-Behmel Síndrome de Beckwith-Wiedemann |
|
| visceromegalias | Síndrome de Beckwith-Wiedemann Síndrome de Simpson-Golabi-Behmel |
|
| Idade Óssea avançada | Síndrome de Sotos Síndrome de Weaver Síndrome de Nevo |
|
| desproporcional | inteligência normal | Síndrome de Marfan Hipogonadismo |
| desporporcional | déficit intelectual | Síndrome de Klinefelter Síndrome XYY Homocistinúria |
| Fonte: modificado de ALVES, 2019. | ||
| SÍNDROME | INÍCIO PRÉ-NATAL | HIPOGLICEMIA NEONATAL | MACROCRANIA / DOLICOCEFALIA | CARDIOPATIA | NEFROPATIA | PROPORÇÃO EUNUCÓIDE | RISCO DE NEOPLASIA |
|---|---|---|---|---|---|---|---|
| Bannayan-Riley-Ruvalcaba | + | ||||||
| Beals | + | + | |||||
| Beckwith-Wiedemann | + | + | + | + | + | ||
| Costello | + | + | |||||
| Elejalde | + | + | + | ||||
| Klinefelter | + | ||||||
| Marfan | + | + | |||||
| Marshall-Smith | + | + | |||||
| Nevo | + | + | |||||
| Pallister-Killian | + | + | |||||
| Partington | + | ||||||
| Perlman | + | + | |||||
| Simpson-Golabi-Behmel | + | + | + | + | + | + | |
| Sotos | + | + | + | + | + | ||
| Weaver | + | + | + | ||||
| Fonte: ALVES, 2019. | |||||||
DIAGNÓSTICO LABORATORIAL
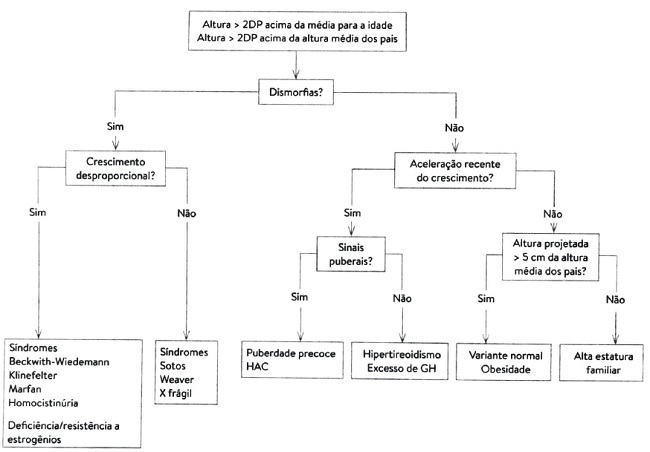
Figura 1: Algoritmo diagnóstico da alta estatura. (Fonte: BESERRA, 2019)
| EXAME | INDICAÇÃO | COMENTÁRIO |
|---|---|---|
| Glicemia Insulina |
Detectar hipoglicemia nos RN filhos de mães diabéticas, hiperinsulinismo neonatal e síndromes genéticas (Beckwith-Wiedemann; Simpson-Golabi-Behmel, Perlman)1. | |
| TSH T3 T4L |
Detectar hipertireoidismo1. | |
| GH basal | GH basal < 0,4 ng/mL torna pouco provável o diagnóstico de adenomas secretores de GH1. | |
| IGF-1 IGFBP-3 |
Investigar hipersecreção de GH1. | |
| Prolactina | Investigar turmores produtores de GH1. | |
| LH FSH Testosterona ou estradiol |
pacientes com atraso ou avanço puberal1. | Investigar hipogonadismo ou puberdade precoce1. |
| Cortisol sérico matinal ACTH |
|
|
| Homocisteína sérica e urinária | Investigar homocistinúria1. | |
| Cariótipo | suspeita de síndrome genética, principalmente meninos com relaçao segmento superior/segmento inferior reduzida ou retardo mental1. | Investigar alta estatura sindrômica1. |
| Idade Óssea | ||
| Raios-X de tórax (PA e P) | suspeita de síndrome genética1. | |
| USG de abdome total | suspeita de síndrome genética1. | Detectar tumores (tumor de Wilms, hepatoblastoma, etc) e visceromegalias1. |
| Ecocardiograma | suspeita de síndrome genética1. | |
| Avaliação Oftalmológica | suspeita de síndrome de Marfan e Homocistinúria1. |
|
| EXAME | INDICAÇÃO | COMENTÁRIO |
|---|---|---|
| Dosagem de GH após Teste de Tolerância Oral à Glicose (TTOG) | Método: administrar 1,75 g/Kg de glicose (máximo: 75g), VO, e dosar GH sérico nos tempos 0, 30, 60, 90 e 120 minutos1. Resposta:
Observação: pacientes adolescentes podem apresentar resultado falso-positivo, não havendo supressãodo GH após o estímulo1.
|
|
| RMN sela túrcica | gigantismo, puberdade precoce ou atrasada | |
| Avaliação com geneticista | suspeita de síndrome genética1. | |
| EXAME | SÍNDROME GENÉTICA |
|---|---|
| Sequenciamento do gene GCP3 | Síndrome de Simpson-Golabi-Behmel |
| FISH para o cromossomo 5q35 | Síndrome de Sotos |
| FISH para o cromossomo 12p | Síndrome de Pallister-Killian |
| FISH para o cromossomo 11p15 | Síndrome de Beckwith-Wiedemann |
| Comparative genomic hybridization (CGH) | |
| Whole exome sequencing (WES) | |
TRATAMENTO
Pacientes com idade óssea superior a 13 anos, se meninas, e superior a 14 anos, se meninos, não se beneficiam em termos de redução da estatura final com o tratamento medicamentoso1.
O método de Greulich-Pyle e de Tanner-Whitehouse Mark 1 e 2 superestimam a previsão da estatura final quando a idade óssea é inferior a 9 anos1.
ALTA ESTATURA CONSTITUCIONAL
- MENINAS:
- ESTRÓGENO:
- ETINILESTRADIOL: 100-300 µg/dia2. Se ocorrer sagramento vaginal ou, após 1 ano do início de tratamento, adiciona-se progesterona cíclica à terapia estrogênica (NERTISTERONA: 5 mg/dia, VO, dop 1º ao 14º dia do mês)2.
- Suspender o medicamento quando a Idade Óssea atingir 14 anos, se surgirem efeitos adversos ou quando a família decide pela suspensão1.
- Discutir o tratamento com os pais:
- redução da estatura final entre 1,2 e 8 cm1.
- Efeitos adversos: ganho de peso, hipertensão, náusea, hiperplasia endometrial, tromboembolismo e redução da futura fertilidade1.
- surgimento de caracteres sexuais secundários e menstruação em meninas ainda pré-púberes1.
- LANREOTIDA:
- Análogo da somatostatina, reduzindo a secreção de GH e IGF-11.
- Posologia:30 mg, IM, a cada 10 a 14 dias, pelo tempo médio de 22 meses1.
- Reduz a estatura final em 3,8 cm1.
- Efeitos adversos: diarréia, vômito, cólica abdominal, lama e litíase biliar1.
- ESTRÓGENO:
- MENINOS:
- ENANTADO DE TESTOSTERONA: 250 mg, IM, 15/15 dias1,2.
- Suspender o tratamento quando a Idade Óssea atingir 14 anos, quando surgirem efeitos adversos ou quando a família opta pela suspensão1.
- Discutir o tratamento com os pais:
- redução da estatura final entre 1,2 e 8 cm1.
- Efeitos adversos: início da puberdade em crianças pré-púberes, acne, edema de extremidades, ginecomastia, ereções prolongadas e/ou dolorosas, recessão temporal dos cabelos, etc1.
- falta da acurácia do atlas de Bayley-Pinneau em predizer a estatura final, pois tende a superestimá-la1.
ADENOMA HIPOFISÁRIO SECRETOR DE GH
- 1º Opção: TRATAMENTO CIRÚRGICO:
- Remoção dos tumores pequenos e bem delimitados por neurocirurgia (preferencialmente transesfenoidal)1.
- Sucesso cirúrgico:
- GH após TTOG < 1 ng/mL e redução dos níveis sérios de IGF-1 e IGFBP-31.
- Complicações cirúrgicas: hipopituitarismo, fistula liquórica, lesão do nervo óptico, meningite, etc1.
- 2º Opção: TRATAMENTO MEDICAMENTOSO:
- ANÁLOGOS DA SOMATOSTATINA:
- Suprimem a secreção de GH e IGF-1, mas são pouco eficazes na redução tumoral1.
- OCTREOTIDA (SANDOSTATIN: ampolas de 0,05, 0,1 e 0,5 mg):1 a 40 mg/Kg/dia, SC, 8/8 horas1.
- OCTREOTIDA DE LONGA DURAÇÃO (SANDOSTATIN LAR: ampolas de 30 mg): 10 a 30 mg, IM, a cada mês1.
- LANREOTIDA (SOMATULINE LP: ampola 30 mg): 30 mg, IM, a cada 7 a 14 dias1.
- Efeitos colaterais: vômito, diarréia, dor abdominal, flatulência, colelitíase (20-30%), hipo ou hiperglicemia, queda de cabelos, bradicardia sinusal e hipotireoidismo central1.
- AGONISTAS DA DOPAMINA:
- Reduzem pouco a secreção de GH, sendo mais indicados para os casos com cossecreção de GH e prolactina pelo adenoma hipofisário1.
- BROMOCRIPTINA (PARLODEL: comprimido 2,5 e 5 mg): iniciar com 1,25 mg, VO, ao deitar e aumentar progressivamente, se necessário, até 5 a 7,5 mg/dia, VO, a cada 8 horas1.
- CABERGOLINA (DOSTINEX: comprimido 0,5 mg): iniciar com 0,25 mg, VO, 1 vez por semana e aumentar, se necessário, até 3,5 mg/semana, VO, em 1 ou 2 doses semanais1.
- Efeitos colaterais: cefaléia, tontura, hipotensão ortostática, contestão nasal, constipação, exacerbação e distúrbios do humor (menos frequentes com a cabergolina), calcificação aórtica, regurgitação tricúspide e fibrose pulmonar e retroperitoneal1.
- Monitoriar com ecocardiogramas periódicos1.
- BLOQUEADORES (ANTAGONISTAS) DO RECEPTOR DE GH:
- Indicado para os casos que não respondem aos análogos de somatostatina1. Pode ser associado a outros medicamentos1.
- PEGVISOMANTO (SOMAVERT: ampola 10, 15 e 20 mg): 10 a 40 mg, SC, 1 vez ao dia1. Fazer rodízio dos locais de aplicação1.
- Efeitos colaterais: hipertrofia nos locais de aplicação e elevação das transaminases1.
- ANÁLOGOS DA SOMATOSTATINA:
- 3° Opção: RADIOTERAPIA: indicada para os casos que não podem ser submetidos ao tratamento cirúrgico, ou com resultado insatisfatório a este e que não tolerem o tratamento medicamentoso2.
MONITORIZAÇÃO DE NEOPLASIAS
| SÍNDROME | NEOPLASIAS |
|---|---|
| Realizar USG de abdome, Hemograma completo e dosagem do antígeno carcinoembrionário periodicamente: | |
| Beckwith-Wiedemann | Tumor de Wilms (0-8 anos de idade2), nefroblastoma, hepatoblastoma (0-4 anos de idade2), neuroblastoma, rabdomiossarcoma, tumor do córtex adrenal. USG ABDOMINAL e ALFAFETOPROTEÍNA a cada 3 meses até 4 anos de idade e, a partir de 4 anos, USG ABDOMINAL a cada 4 meses até 8 anos de idade)2. |
| Sotos | Tumor de Wilms, carcinoma hepático, vaginal e das parótidas e tumores neuroendócrinos |
| Weaver | Neuroblastoma |
| Perlman | Tumor de Wilms |
| Simpson-Golabi-Behmel | Tumor de Wilms, hepatoblastoma, gonadoblastoma, neuroblastoma, carcinoma hepatocelular |
| Bannayan-Riley-Ruvalcaba | Seminoma, germinoma |
| Fonte: modificado de ALVES, 2019. | |
REFERÊNCIAS BIBLIOGRÁFICAS
- ALVES, C.A.D.: Alta estatura . In: _______: Endocrinologia Pediátrica. Barueri-SP, Editora Manole, 1° edição, 2019: 27-44.
- BESERRA, I.C.R.: Alta estatura. In: MADEIRA, I.R. & CORDEIRO, M.M.: Endocrinologia Pediátrica. Barueri-SP, Editora manole, 2º ediçao, 2019: 9-23.